Cholesterol Synthesis
As an essential component of cellular membranes and a precursor for the synthesis of all steroid hormones, as well as bile acids and vitamin D, cholesterol plays a pivotal role in many physiological functions. However, excess levels of plasma cholesterol can become toxic and are directly related to the risk for CV events.1,2
The liver is the main site for the production of cholesterol as well as for the regulation of serum low-density lipoprotein cholesterol (LDL-C) levels through LDL receptor (LDLR)-mediated clearance.3 In addition to LDL produced by the liver, cholesterol is obtained through dietary sources and produced de novo by other tissues (eg, CNS and steroidogenic tissues).4,5 While systemic LDL-C can be an important source of cholesterol, all mammalian cells have the ability to synthesize it de novo from acetate.6
LDLR Recycling and Cholesterol Homeostasis
Both dietary and de novo produced cholesterol is transported in the blood by apolipoprotein B (apoB)-containing lipoproteins, including cholesterol-rich LDL.1 LDL uptake occurs through binding of apoB-containing lipoproteins to the LDLR and internalization of the complex through endocytosis. After internalization, the receptors dissociate from their ligands and are recycled back to the surface while LDL is hydrolyzed in lysosomes.1
An estimated 70% of systemic LDL-C is cleared from the circulation through the LDLR, while the remaining 30% is eliminated through intracellular scavenger receptors. Homeostatic control of intracellular cholesterol levels is achieved through complex regulatory feedback loops that ensure intracellular levels remain constant regardless of changes in circulating LDL-C levels.1 Early studies in patients with genetic disorders of lipid metabolism demonstrated how intracellular cholesterol levels regulate both the rate-limiting step in the cholesterol biosynthetic pathway and the number of LDLRs responsible for systemic LDL-C uptake to balance de novo synthesis and uptake.1
Recycling of LDLRs Enables Efficient Clearance of LDL Particles1,7,8

Regulation by PCSK9
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a member of the mammalian proprotein convertase family of secretory serine endoproteases that functions as a molecular chaperone, binding to LDLR and targeting it for lysosomal degradation.9,10 PCSK9 directs the degradation of LDLR via two separate pathways:
- In the intracellular pathway, nascent PCSK9 binds to LDLR and directs it from the trans-Golgi to the lysosome.9
- In the extracellular pathway, secreted PCSK9 binds to LDLR and upon internalization directs it for degradation by the lysosome thus inhibiting further LDLR recycling and LDL-C clearance.9
Resources
View the video below and download the associated materials relating to the role of PCSK9 in the regulation of LDL-C here.

The Role of PCSK9 in the Regulation of LDL-C
Watch the video below, which gives an overview of the role of PCSK9 in cholesterol homeostasis through regulation of LDLR degradation.
Genetic Variations in PCSK9: Gain-of-function and Loss-of-function Variants9,11
Gain-of-function mutations in PCSK9 were first identified in French and then shortly after in Norwegian families with clinical familial hypercholesterolemia (FH) and were shown to be responsible for the hyperlipidemia phenotype.12,13 In contrast, loss-of-function mutations in PCSK9 were associated with significant reductions in circulating LDL-C levels.12 Case studies of individuals with loss-of-function PCSK9 mutations reveal that long-term exposure to very low LDL-C levels (14–16 mg/dL) is not associated with any obvious adverse effects.14-17
Genetic Variants of PCSK9 Demonstrate Its Importance in Regulating LDL Levels7,11,18
Drag the bar across the image below to see the difference in gain- or loss-of-function mutations.
Gain-of-function
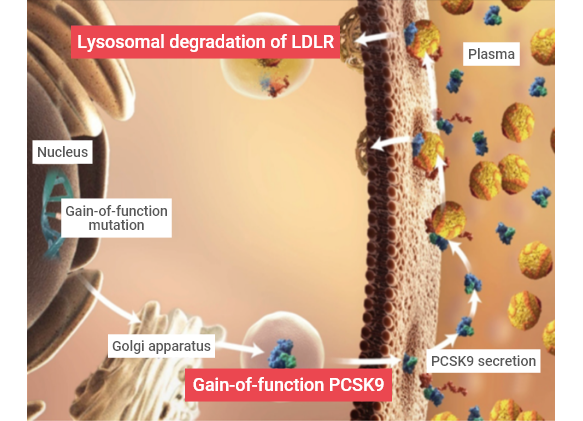
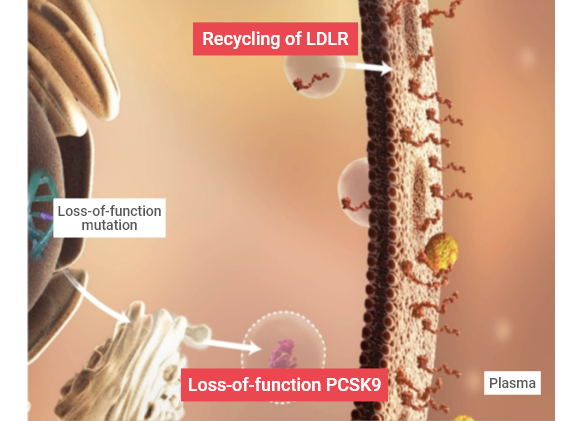
Loss-of-function
- Goldstein JL, Brown MS. History of discovery: the LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431-438. doi:10.1161/ ATVBAHA.108.179564.
- Lagor WR, Millar JS. Overview of the LDL receptor: relevance to cholesterol metabolism and future approaches for the treatment of coronary heart disease. Journal of Receptor, Ligand and Channel Research. 2010;3:1-14. doi:10.2147/JRLCR.S6033.
- Turley SD. Cholesterol metabolism and therapeutic targets: rationale for targeting multiple metabolic pathways. Clin Cardiol. 2004;27(suppl 3):16-21. doi:10.1002/clc.4960271506.
- Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24(5):806-815. doi:10.1161/01.ATV.0000120374.59826.1b.
- Katsuno M, Adachi H, Sobue G. Getting a handle on Huntington’s disease: the case for cholesterol. Nat Med. 2009;15(3):253-254. doi:10.1038/nm0309-253.
- Morales-Villegas EC, Ray KK. Physiological level of LDL cholesterol: the master key a novel dream comes true. Cardiovasc Pharm Open Access. 2016;6:5. doi:10.4172/2329-6607.1000223.
- Steinberg D, Witztum JL. Inhibition of PCSK9: a powerful weapon for achieving ideal LDL cholesterol levels. Proc Natl Acad Sci USA. 2009;106(24):9546-9547. doi:10.1073/pnas.0904560106.
- Brown WM, George M, Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci USA. 1979;76(4):1967-1971. doi:10.1073/ pnas.76.4.1967.
- Lagace TA. PCSK9 and LDLR degradation: regulatory mechanisms in circulation and in cells. Curr Opin Lipidol. 2014;25(5):387-393. doi:10.1097/MOL.0000000000000114.
- Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114(6):1022-1036. doi:10.1161/ CIRCRESAHA.114.301621.
- Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264-1272. doi:10.1056/NEJMoa054013.
- Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154-156. doi:10.1038/ng1161.
- Leren TP. Mutations in the PCSK9 gene in Norwegian subjects with autosomal dominant hypercholesterolemia. Clin Genet. 2004;65(5):419-422. doi:10.1111/j.0009-9163.2004.0238.x.
- Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79(3):514-523. doi:10.1086/507488.
- Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193(2):445-448. doi:10.1016/j.atherosclerosis.2006.08.039.
- Cariou B, Ouguerram K, Zaïr Y, et al. PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2009;29(12):2191-2197. doi:10.1161/ATVBAHA.109.194191.
- Olsson AG, Angelin B, Assmann G, et al. Can LDL cholesterol be too low? Possible risks of extremely low levels. J Intern Med. 2017;281(6):534-553. doi:10.1111/joim.12614.
- Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybjaerg-Hansen A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J Am Coll Cardiol. 2010;55(25):2833-2842. doi:10.1016/j.jacc.2010.02.044.
LDL-C Exposure Causes Atherosclerosis and Is a Key Modifiable Factor for the Development of ASCVD1,2
The pathophysiology of atherosclerosis is characterized by the accumulation and retention of cholesterol-rich lipoproteins in the arterial wall, endothelial dysfunction, and a subsequent inflammatory response that results in the formation of atherosclerotic plaques.3,4 The subendothelial retention of cholesterol-rich LDLs and other apolipoprotein B-containing lipoproteins, particularly at sites of disturbed blood flow such as branch points, represents the key initiating step in the atherosclerotic process.1,3-6
Cholesterol-rich LDLs can normally diffuse through the arterial intima.1 At low LDL-C concentrations, typical of newborns (LDL-C ~ 20–40 mg/dL), the probability of LDL particle retention in the arterial wall is low.1 However, as the concentration of LDL-C rises, the probability of retention and the risk for the development of atherosclerosis increases in a dose-dependent manner.1
The accumulation and subsequent oxidation of LDLs trigger a chronic maladaptive inflammatory response by activated endothelial cells, smooth muscle cells and infiltrating macrophages, ultimately leading to the development of atherosclerotic plaques.3,5,7
While the clinical manifestations of atherosclerosis (such as MI and stroke) occur suddenly, the underlying lesions that lead to CV events develop over decades.5,8 Epidemiological studies have shown that prolonged hyperlipidemia increases the risk for CV events.9
The Effect of Cumulative Exposure to LDL on Plaque Burden and Risk of Cardiovascular Disease10
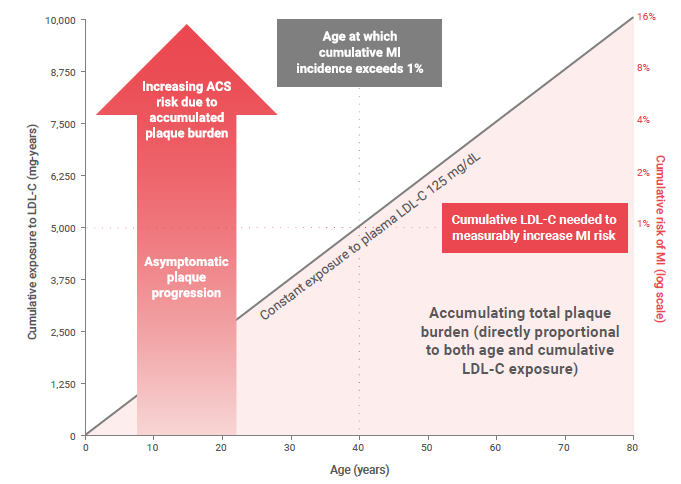
ACS, acute coronary syndrome; LDL, low-density lipoprotein; LDL-C, LDL cholesterol; MI, myocardial infarction.
- Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459-2472. doi:10.1093/eurheartj/ehx144.
- Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):937-952. doi:10.1016/S0140-6736(04)17018-9.
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685-1695. doi:10.1056/NEJMra043430.
- Singh RB, Mengi SA, Xu YJ, Arneja AS, Dhalla NS. Pathogenesis of atherosclerosis: a multifactorial process. Exp Clin Cardiol. 2002;7(1):40-53.
- Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832-1844. doi:10.1161/CIRCULATIONAHA.106.676890.
- Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161(1):161-172. doi:10.1016/ j.cell.2015.01.036.
- Maiolino G, Rossitto G, Caielli P, Bisogni V, Rossi GP, Calò LA. The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediators Inflamm. 2013;2013:714653. doi:10.1155/2013/714653.
- Hong YM. Atherosclerotic cardiovascular disease beginning in childhood. Korean Circ J. 2010;40(1):1-9. doi:10.4070/kcj.2010.40.1.1.
- Navar-Boggan AM, Peterson ED, D’Agostino RB, Neely B, Sniderman AD, Pencina MJ. Hyperlipidemia in early adulthood increases long-term risk of coronary heart disease. Circulation. 2015;131(5):451-458. doi:10.1161/CIRCULATIONAHA.114.012477.
- Ference BA, Graham I, Tokgozoglu L, Catapano AL. Reprint of: impact of lipids on cardiovascular health.J Am Coll Cardiol. 2018;72(23):1141-1156. doi: 10.1016/j.jacc.2018.06.046.